2.5 Der 2. und 3. Hauptsatz der Thermodynamik
- Spontane Prozesse und Reaktionen
Definitionen: Chemische Reaktionen oder physikalische Umwandlungen, die
von selbst, ohne Einwirkung von außen, ablaufen; alle spontanen Prozesse sind
irreversibel, d.h. nicht durch infinitesimal kleine (z.B. dq, dw) Störungen
umkehrbar.
Beispiele:
- „Expansion eines Gases ins Vakuum“

Eine Umkehrung ist nie beobachtet worden, obwohl es nicht dem
1.Hauptsatz widersprechen würde. Denn bei einem idealen Gas gilt:
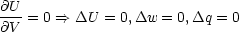
- Chemische Reaktionen
- z.B. 2 H2(g) + O2(g)
 2 H2O(g)
2 H2O(g)
- z.B. Cl-(aq) + Ag+(aq) AgCl(s)
- Wärmeausgleich zwischen zwei Festkörpern:
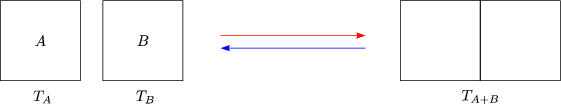
Eine Umkehrung wurde nicht beobachtet, obwohl sie wieder mit
1.Hauptsatz vereinbar wäre. Die Begründung ist folgende:
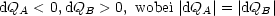
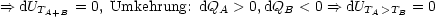
Zusammenfassung:
- Alle spontanen Prozesse laufen in bestimmter Richtung ab.
- Richtung ist nicht mit dem 1.Hauptsatz vorhersagbar, oder mit
anderen Worten: 1.Hauptsatz läßt Umkehrprozesse zu.
Problem:
|
| |
| Wir müssen Zustandsfunktion S = S(V,T) finden, die Richtung spontaner Prozesse angibt. Diese
Größe bezeichnen wir Entropie. |
| |
|
| |
Aufgabenstellung:
- Definition von S
- Zustandsfunktion!
- Zusammenfassung von S = S(V,T) und z.B. U(V,T)
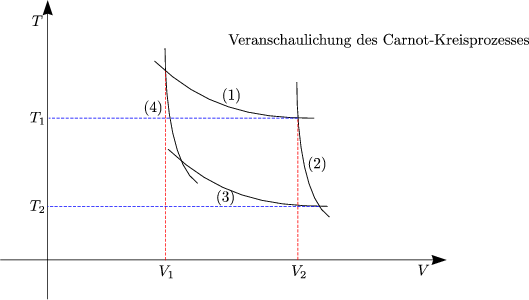
- Carnot-Kreisprozeß:
Wir betrachten eine Wärmekraftmaschine (Carnot-Maschine):
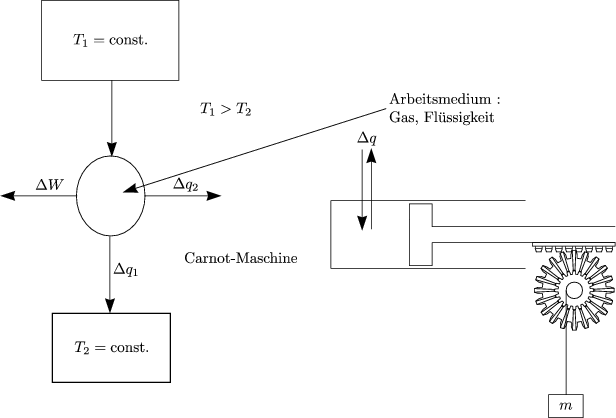
Carnot-Prozeß in einem pV -Diagramm:

Problem:
|
| |
Wie groß ist der Wirkungsgrad 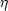 einer Carnot-Maschine? einer Carnot-Maschine? |
Wobei: = 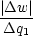 |
| |
|
| |
Lösung:
|
|
|
|
|
| Gas | Schritt | Reservoir: T1 | Reservoir: T2 | Arbeitssumme |
| | | | | hierzu: ideales Gas |
|
|
|
|
|
|
|
|
|
|
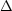 q1 q1 | | | | |
w1 = -nRT1 ln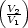 | (1) | -q1 | | -nRT1 ln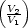 |
| U1 = 0 | | | | |
|
|
|
|
|
| q2 = 0 | | | | |
| w2 = -nCV (T1 - T2) | (2) | | | -nCV (T1 - T2) |
| U2 = w2 | | | | |
|
|
|
|
|
| q3 | | | | |
w3 = nRT2 ln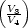 | (3) | | +q3 | nRT2 ln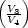 |
| U3 = 0 | | | | |
|
|
|
|
|
| q4 = 0 | | | | |
| w4 = nCV (T1 - T2) | (4) | | | nCV (T1 - T2) |
| U4 = w4 | | | | |
|
|
|
|
|
| |
Bilanz:
- U =
 i = 14Ui
i = 14Ui  0 = 0 - nCV (T1 - T2) + 0 + nCV (T1 - T2) = 0
0 = 0 - nCV (T1 - T2) + 0 + nCV (T1 - T2) = 0
Das heißt, daß U eine Zustandsfunktion ist, oder mit anderen Worten,
daß das Ringintegral für einen
Umlauf gleich 0 ist:
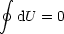
- w =
i = 14wi = -nRT1 ln
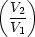 - nCV (T1 - T2) + nRT2 ln
- nCV (T1 - T2) + nRT2 ln + nCV (T1 - T2)
+ nCV (T1 - T2)

Auf der Suche nach Entropiefunktion S = S(V,T) betrachten wir den
Carnot-Kreisprozeß:

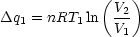
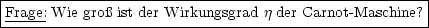
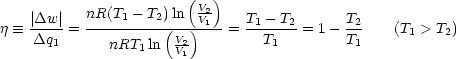
Fazit:

Anmerkung:
__________ Dies gilt nicht nur für ein ideales Gas als Arbeitsmedium, sondern
allgemein. Man sollte sich merken, daß steigt, wenn T1 erhöht wird.
2.5.1 Entropie und der 2.Hauptsatz
Suche Funktion S(V,T), welche die Richtung
angibt, in der spontane und damit irreversible Prozesse verlaufen.
Lösung:
|
| |
Wir gehen vom Carnot-Kreisprozeß aus: = 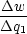 = = 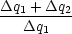 = =  |
Denn: q1 = nRT1 ln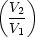 ; q2 = -nRT2 ln ; q2 = -nRT2 ln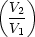 |
| |
|
| |
Damit gilt also:

Interpretation: Die Summe der reduzierten Wärmen  ist für reversible
Kreisprozesse 0:
ist für reversible
Kreisprozesse 0:
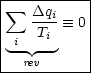
Ein beliebiger reversibler Kreisprozeß kann durch infinitesimal kleine
Carnot-Kreisprozesse approximiert werden.

Daraus ergibt sich folgender Grenzwert:
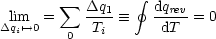
Die Funktion 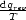 hat damit die Eigenschaften einer Zustandsfunktion. Dies führt
zur thermodynamischen Definition von Entropie:
hat damit die Eigenschaften einer Zustandsfunktion. Dies führt
zur thermodynamischen Definition von Entropie:
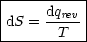
Frage:
|
| |
| Gibt S auch die Richtung spontaner irreversibler Prozesse an? |
| |
|
| |
Antwort:
|
| |
| Wir betrachten einen modifizierten Carnot-Prozeß mit 2 irreversiblen Schritten (2) und (4). |
| |
|
| |
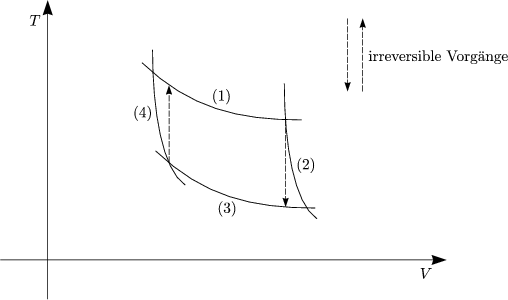
Für einen Kreisprozeß mit irreversiblen Teilschritten gilt:
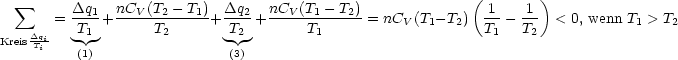
Zusammenfassung:
Wir zerlegen den irreversiblen Prozeß in infinitesimal kleine
Teilschritte - analog vorher -
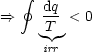
Wir betrachten folgenden Kreisprozeß:
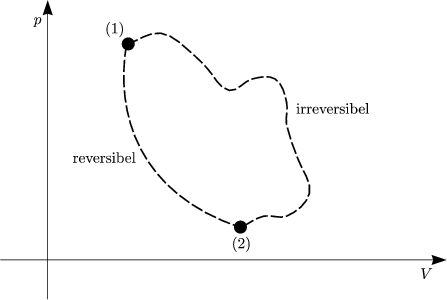
Da der 1.Teilschritt irreversibel ist, gilt:
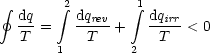
In reversiblen Teilschritten resultiert:

Wir erhalten für den gesamten Kreisprozeß:
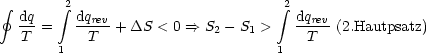
2.Hauptsatz:
Zustandsänderungen in einem isolierten System haben folgende Entropieänderungen
zur Folge:
- Wenn reversibel: S = 0
- Wenn irreversibel: S > 0
Die Entropie gibt somit die Richtung irreversibler Prozesse an. Ein solcher Prozeß
liegt genau dann vor, wenn S > 0 gilt. Wir fassen unsere Ergebnisse zu S = S(V,T)
bzw. S = S(p,T) zusammen:
- Wirkungsgrad einer Carnot-Maschine:
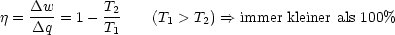
- dS
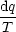 bei reversibler Zustandsänderung
bei reversibler Zustandsänderung
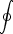 dS = 0 d.h. Zustandsfunktion
dS = 0 d.h. Zustandsfunktion
- In einem abgeschlossenen System gilt:
S S2 - S1 0 für reversible Prozesse, S = S2 - S1 > 0 für
irreversible Prozesse
2.5.2 Statistische Interpretation der Entropie
Einführendes Beispiel: spontaner Prozeß

Nach dem 2.Hauptsatz gilt S2 - S1 > 0. Fakt ist, daß Zustand (2) ungeordneter als
Zustand (1) ist. Molekeln in (2) haben mehr Möglichkeiten, sich in V 2 zu
verteilen, also mehr Realisierungsmöglichkeiten. Dieser Zustand ist folglich viel
wahrscheinlicher!
Vermutung:
Frage:
|
| |
Was ist der Zusammenhang zwischen S und der Unordnung eines Zustandes? Oder mit anderen Worten:
S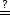 f(N)? f(N)? |
| |
|
| |
Lösung:
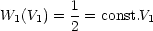
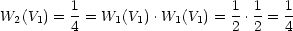
Daraus folgt WN(V 1) = W1(V 1)N. Die Wahrscheinlichkeiten sind multiplikativ
verknüpft. Kommen wir nun zurück zu f(W)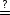 S. S ist eine extensive
thermodynamische Variable, d.h. S2N = SN + SN. Wir suchen also eine Funktion,
durch die:
S. S ist eine extensive
thermodynamische Variable, d.h. S2N = SN + SN. Wir suchen also eine Funktion,
durch die:
- Multiplikative Eigenschaft von W(V ) berücksichtigt wird
- Additive Verknüpfung bei S berücksichtigt wird
Dies erfüllt gerade der Logarithmus:
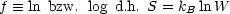
Frage:
|
| |
| Warum handelt es sich bei der Konstanten vor dem Logarithmus um die BOLTZMANNkonstante kB? |
| |
|
| |
Antwort:
|
| |
| Wir betrachten die isotherme reversible Expansion eines idealen Gases: |
| |
| dU = dq + dw = dq - pdV = CV dT - pdV = 0 |
| |
dqrev = -dw = pdV 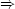 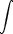 (1)(2)
(1)(2)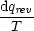 =
(1)(2) =
(1)(2)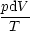 =
(1)(2) =
(1)(2)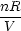 dV = R ln dV = R ln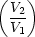 |
| |
S = R ln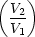 = NA . kB ln = NA . kB ln = kB ln = kB ln NA NA
|
| |
|
| |
2.5.3 Entropieänderungen bei einfachen reversiblen Zustandsänderungen
Beispiel: Tiefenabhängigkeit bei p=const. oder V =const.

Wir betrachten den Fall V =const.:
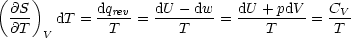
Es resultiert hiermit:
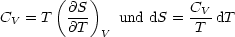
Daraus folgt wiederum die Beziehung:

2.5.4 Umwandlungsentropien
Wir betrachten einen reinen Stoff und wollen wissen, wie Phasenumwandlungen und
Entropieänderung S zusammenhängen:

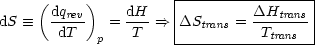
Nach der TROUTONschen Regel gilt näherungsweise:
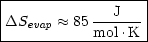
2.5.5 Absolutberechnung der Entropie einer Substanz und 3.Hautpsatz der
Thermodynamik
Frage:
|
| |
Wie groß ist S(T), ausgehend von T = 0 T bei p =const.? T bei p =const.? |
| |
|
| |
Antwort:
|
| |
S(t) = S(T = 0) +
0TSm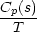 dT + dT + 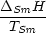 +
TSmTS +
TSmTS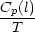 dT + dT + 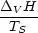 +
TST +
TST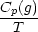 dT dT |
| |
|
| |
Man wird sich nun fragen, wie groß die Entropie beim absoluten Nullpunkt ist:
S(T = 0) =? Dazu kühlen wir im Gedankenexperiment einen Festkörper langsam ab,
so daß ein perfekter Kristall vorliegt.
3.Hautpsatz:
|
| |
Alle perfekten (im inneren thermischen Gleichgewicht befindlichen) Kristalle haben dieselbe Entropie
S(T = 0), d.h. S 0 für T 0 für T 0. Diese Aussage ist mit BOLTZMANN konsistent. 0. Diese Aussage ist mit BOLTZMANN konsistent. |
| |
|
| |