Diese tritt bei abgeschlossenen Elektronenschalen auf. Wichtig ist sie beispielsweise bei Edelgasen, Molekülen (wie H2) und Polymeren (Kohlenwasserstoff). Dieser Bindungstyp tritt immer auf; er liefert einen kleinen Zusatzbeitrag bei anderen Bindungstypen. Die Van-der-Waals-Wechselwirkung ist eine Dipol-Dipol-Wechselwirkung.
Edelgasatome besitzen kugelförmige Wellenfunktionen. Nichts desto trotz liegen fluktuierende Dipole p1 vor, obwohl <p1> = 0 ist. Aufgrund dieses fluktuierenden Dipols entsteht in dessen Umgebung ein elektrisches Feld. Dieses besitzt folgende Eigenschaft:
Dieses elektrische Feld erzeugt dann in den Nachbaratomen wieder ein
elektrisches Feld, für das p2 = ![]() E (
E (![]()
![]() ) gilt, wobei
) gilt, wobei ![]() die Polarisierbarkeit ist.
Damit gilt:
die Polarisierbarkeit ist.
Damit gilt:
Man benutzt dann folgende Beziehung:
Die Anziehung fällt infolgedessen sehr rasch für große r ab. Daraus folgt der niedrige Schmelzpunkt. Das Lennard-Jones-Potential ergibt sich nun durch Zusammensetzung von anziehendem und abstoßendem Potential:
Die Potentialformel wird oft so genutzt, wie letztlich angeschrieben.
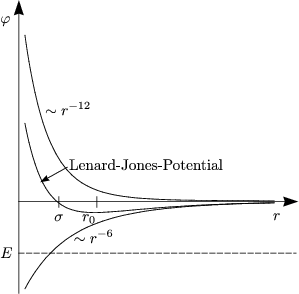
Das Minimum befindet sich bei r0 = 1,12![]() . Wir notieren uns einige
Lennard-Jones-Parameter:
. Wir notieren uns einige
Lennard-Jones-Parameter:
| Element | Ne | Ar | Kr | Xe |
| | 2,74 | 3,4 | 3,65 | 3,98 |
| E [meV] | 3,1 | 10,4 | 14,1 | 20 |
Man kann die Gitterenergie abschätzen, ohne die Nullpunktsenergie zu
berücksichtigen. Das Verfahren ist jedoch sicher ungenau für flüssiges He für
T![]() 0 (fest bei Druck > 2,5MPa). Die Bindungsenergie für ein Atom m ergibt
sich nun durch Summation der Energien bezüglich aller anderen Atome n im
Abstand rmn:
0 (fest bei Druck > 2,5MPa). Die Bindungsenergie für ein Atom m ergibt
sich nun durch Summation der Energien bezüglich aller anderen Atome n im
Abstand rmn:
Der Faktor ![]() kommt daher, weil man alle Energien zweifach summiert. Bei
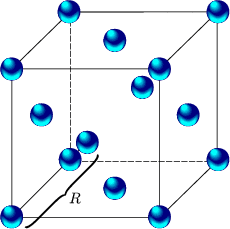
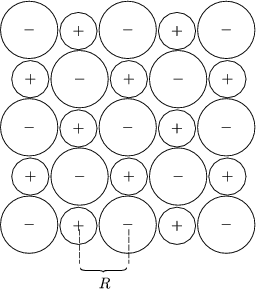
Edelgasen liegt ein kubisch flächenzentriertes Gitter vor:
kommt daher, weil man alle Energien zweifach summiert. Bei
Edelgasen liegt ein kubisch flächenzentriertes Gitter vor:
| 12 | Abstand R |
| 6 | Abstand R |
| | |
Damit folgt die Bindungsenergie:
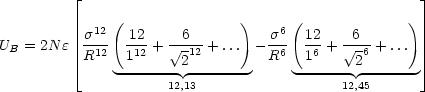
Der Gleichgewichtsabstand R0 erhält man durch Berechnung des Minimums der Wechselwirkungsenergie UB:
Es ergibt sich dann R0 = 1,09![]() und UB(R0) = -8,60N
und UB(R0) = -8,60N![]() .
.
| Element | Ne | Ar | Kr | Xe |
| | 1,15 | 1,11 | 1,09 | 1,09 |
Abweichungen sind erklärbar durch Nullpunktsschwingungen. UB(R0) stimmt gut und sehr gut unter Berücksichtigung der Nullpunktsschwingungen. Siehe hierzu auch Isotopeneffekt:
Eine heteropolare Bindung kann entstehen durch Elektronentransfer wie beispielsweise bei Na+ und Cl-. Infolge der Coulombkräfte entsteht eine ungerichtete Bindung. Für ein Ionenpaar bestehend aus kugelförmigen Ionen wollen wir eine erste Abschätzung vornehmen:
Bei mehreren Nachbarn ist die Energie eher größer. Diese Energie kann man experimentell nur indirekt messen, weil freie Ionen nicht existieren. Hierzu verwendet man einen erdachten Kreisprozeß, nämlich den sogenannten Born-Haber-Kreisprozeß:
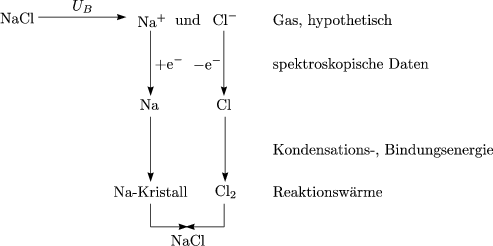
Als Bilanz ergibt sich UB = -8,16eV/Paar. Wir wollen die Bindungsenergie berechnen:
Das Potential für ein Ion m ergibt sich durch:
Der erste Term berücksichtigt die Abstoßung und der zweite die Coulomb-Wechselwirkung. Berücksichtigen wir nur die Anzahl z der nächsten Nachbarn, so gilt folgende Näherung:
Der zweite Term konvergiert schlecht, wegen der ![]() -Abhängigkeit. Dieses
Problem löst man durch Einführung der Madelungkonstante, in die man alles,
was bei der Summation gefährlich ist, steckt:
-Abhängigkeit. Dieses
Problem löst man durch Einführung der Madelungkonstante, in die man alles,
was bei der Summation gefährlich ist, steckt:
Diese Konstante enthält die Struktur des Festkörpers. Betrachten wir beispielsweise eine lineare Kette:
![]()
In drei Dimensionen liegt nur „bedingte“ Konvergenz vor. Ein besonderes Verfahren ist die Konstruktion einer Evjen-Zelle. Betrachten wir hierzu ein zweidimensionales Beispiel:
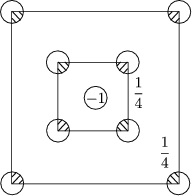
Man konstruiert also ladungsneutrale Zellen. Durch schrittweise Vergrößerung
der Ladungsbruchteile ergibt sich eine schnelle Konvergenz. Als Beispiel in drei
Dimensionen betrachten wir CsCl (8 .![]() Ladung). Die Madelungkonstante ist
strukturabhängig.
Ladung). Die Madelungkonstante ist
strukturabhängig.
| NaCl-Struktur | |
| CsCl-Struktur | |
| ZnS-Struktur | |
Für N Ionenpaare ergibt sich:
Die Gleichgewichtslage erhält man wieder durch Nullsetzen der ersten Ableitung:
Damit resultiert:
Den ersten Term bezeichnet man als Madelungsenergie. Der zweite Term ist
ein Korrekturterm (Abstoßung ![]() 10% bei R0). Für NaCl mit R0 = 2,82Å
folgt:
10% bei R0). Für NaCl mit R0 = 2,82Å
folgt:
Aus der Kompressibilität ergibt sich, daß die Abstoßung bei R = R0 weniger
steil ist als ![]() . Besser ist hier das Born-Mayer-Potential:
. Besser ist hier das Born-Mayer-Potential: