Es liegen atomare Wellenfunktionen mit einem schwachen Überlapp vor. Die Diskussion dieses Problems funktioniert ähnlich wie bei der kovalenten Bindung. Damals hatten wir das LCAO-Modell (linear combination of atomic orbitals) betrachtet. Wir nehmen also an, daß das atomare Problem gelöst sei:
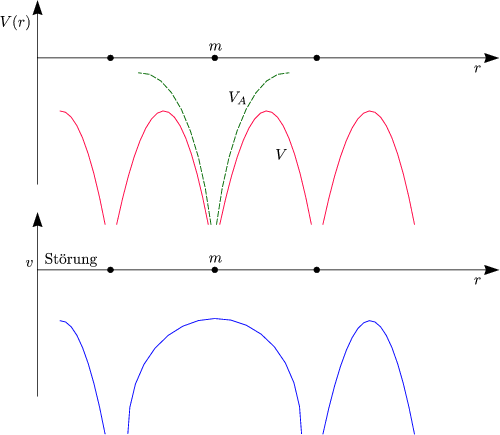
In einem Kristall wollen wir die Einelektronen-Näherung durchführen. Wir betrachten ein Elektron m im Gesamtpotential V = V a + v.
Hierbei handelt es sich um eine Summe über alle anderen Atome. Zu lösen ist dann das Eigenwertproblem:
![]()
![]() (r) müssen bekanntlich wieder Bloch-Wellen sein. Die Energie-Eigenwerte, wenn
wir die Wellenfunktion
(r) müssen bekanntlich wieder Bloch-Wellen sein. Die Energie-Eigenwerte, wenn
wir die Wellenfunktion ![]() k kennen, können berechnet werden durch:
k kennen, können berechnet werden durch:
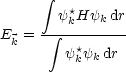
Die Idee ist nun wieder das Ritzsche Variationsverfahren mit einer Versuchsfunktion
![]() k. Dann muß gelten:
k. Dann muß gelten:
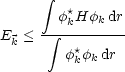
Ek kann als Minimum angenähert werden durch Variation von eben dieser
Versuchsfunktion ![]() . Versuch:
. Versuch: ![]() sei eine lineare Überlagerung der Atomwellenfunktionen
sei eine lineare Überlagerung der Atomwellenfunktionen
![]() (
(![]() -
-![]() ), wobei wir die Vektorpfeile wieder weglassen wollen:
), wobei wir die Vektorpfeile wieder weglassen wollen:
An dieser Stelle nützen wir nun aus, daß es sich um eine Blochwelle handelt, da ja
ein periodisches Gitter vorliegt. Diese Bloch-Welle muß aus einer gitterperiodischen
Modulation und einen Phasenfaktor (Wellenanteil) bestehen. Der gitterperiodische
Anteil muß hierbei in der Summe über die ![]() stecken und der Phasenfaktor in den
am. Wir schreiben also die Versuchsfunktion auf als:
stecken und der Phasenfaktor in den
am. Wir schreiben also die Versuchsfunktion auf als:
Damit können näherungsweise die Energiewerte Ek berechnet werden.
Wir betrachten ![]() als lineare Überlagerung der Atomwellenfunktionen
als lineare Überlagerung der Atomwellenfunktionen ![]() (
(![]() -
-![]() ).
).
Dies muß eine Blochwelle (=gitterperiodische Modulation × Phasenfaktor), also verwenden wir die Versuchsfunktion:
Jetzt berechnen wir Ek näherungsweise. Dazu berechnen wir den Nenner des Bruches:
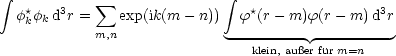
Bei starker Lokalisierung sind nur Glieder mit m = n wichtig und daher kann folgende Näherung benutzen:
Damit erhalten wir:
i ist der Index der atomaren Wellenfunktion. Wendet man HA auf die linke
komplex konjugierte Wellenfunktion ![]() *(r - m) an, so erhält man die atomaren
Eigenwerte Ei für m = n. Wir spalten das ganze in zwei Anteile Ai und Bi
auf:
*(r - m) an, so erhält man die atomaren
Eigenwerte Ei für m = n. Wir spalten das ganze in zwei Anteile Ai und Bi
auf:
Hierbei handelt es sich um die Energieabsenkung mit Bezugselektron m aufgrund von v.
Dies ist ein Überlappintegral vom Bezugselektron m mit Nachbarn n. Durch Summation über m ergibt sich jeweils ein Faktor N:
Pni beschreibt die Geometrie der Umgebung (Gitterstruktur). Die Summe über die
nächsten Nachbarn ist hierbei meist ausreichend. Als Beispiel betrachten wir den
einfachsten Fall, nämlich daß ![]() kugelsymmetrisch ist. Wir betrachten nur die
nächsten Nachbarn eines kubisch primitiven Gitters. Dieses besitzt genau 6 nächste
Nachbarn:
kugelsymmetrisch ist. Wir betrachten nur die
nächsten Nachbarn eines kubisch primitiven Gitters. Dieses besitzt genau 6 nächste
Nachbarn:
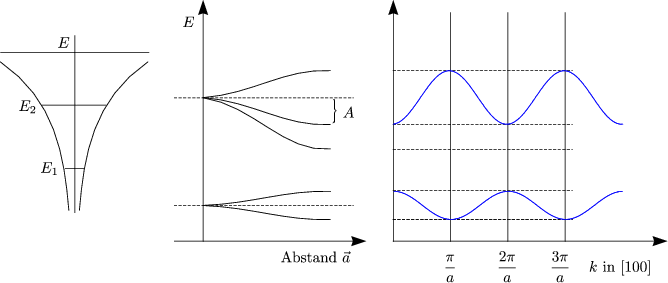
Ai beschreibt die globale Energieabsenkung. Der Bandindex Bi bestimmt die Breite des Bandes. Ai und Bi hängen im allgemeinen von i ab. Betrachten wir Ek bei k = 0:
Bei k = ![]() gilt:
gilt:
Vergleiche dies mit E = ![]() beim freien Elektron. Die effektive Masse
mi* = ±
beim freien Elektron. Die effektive Masse
mi* = ±![]() kann sehr verschieden von m0 sein. Als Beispiel kann man hier den
Ionenkristall K+Cl- anführen. Hier ist der Überlapp klein, weshalb somit die Bänder
schmal sind. Bei kleinem Überlapp ist auch der Faktor Bi klein und damit wird auch
die Breite der Oszillationen klein.
kann sehr verschieden von m0 sein. Als Beispiel kann man hier den
Ionenkristall K+Cl- anführen. Hier ist der Überlapp klein, weshalb somit die Bänder
schmal sind. Bei kleinem Überlapp ist auch der Faktor Bi klein und damit wird auch
die Breite der Oszillationen klein.
Bei kovalenten Bindungen, wie diese beispielsweise im Diamant auftreten, entstehen
aus s- und p-Orbitalen sogenannte sp3-Hybride (im Festkörper). Bei 4 Elektronen pro
Atom im Valenzband erhält man ELücke ![]() 1eV für Si und etwa 5eV für
C.
1eV für Si und etwa 5eV für
C.